Using EnsDb's annotation database in Python
I found that there isn’t a systematic way to query and convert genomic annotation IDs in Python. At least there isn’t one as good as what R/Bioconductor currently has. If you’ve never heard of R/Bioconductor annotation tool stack before, check out the official workflow or my post in 2016 specific for querying Ensembl annotations.
Although I enjoy using R for genomic annotation conversion, a few days ago I wanted to do the same thing inside my text processing script in Python. I might be able to re-write the script in R but I feel like R is not really the right tool for this task and on top of it, I don’t know how to write an efficent text processing in R1.
Knowing the fact that all annotations in R are stored in single-file SQLite databases, I should be able to connect the database directly Python or any other language and wirte SQL query to retrieve the same information. So my question now becomes to how to extract or find the path to the databases. Turn out that many new Bioconductor annotation packages are hosted via AnnotationHub, and user can search for the annotation package and retrieve them locally by their ID. For example, all the recent Ensembl releases, e.g., EnsDb.Hsapiens.vXX, are available on AnnotationHub.
After digging around a bit, I am able to query the AnnotationHub, download the correct EnsDB SQLite database file, and make SQL queries for the annotation ID conversion without any R package. I will share the details in the rest of the post.
But before we start with the details, I want to clarify that it wasn’t my intention to persuade people away from the current R ecosystem. The current R ecosystem is great and I will recommend people to stick with it as much as you can. I am pretty sure I will hit a lot of issues if I want to do more complex analysis or queries without the help of what R packages provide.
AnnotationHub web interface
EDIT 2019-01-29
Now AnnotationHub has a nice web interface. With the new API, we can search and download all the EnsDb annotation objects on AnnotationHub by visiting https://annotationhub.bioconductor.org/package2/AHEnsDbs:
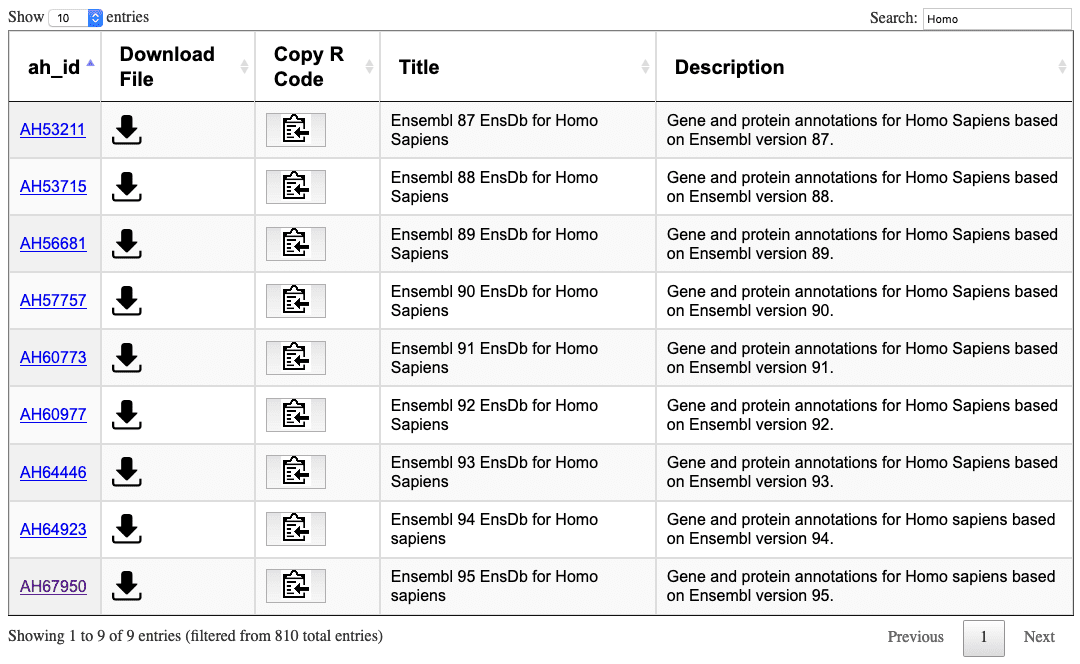
The following section is the old way to navigate through AnnotationHub’s database.
Manual query in AnnotationHub
When one wants to use the R package AnnotationHub, the common usage is
library(AnnotationHub)
ah <- AnnotationHub()
## snapshotDate(): 2017-10-27
query(ah, c("EnsDb", "Homo sapiens"))
The function call AnnotationHub() will download the latest version of the metadata of all available annotation object. The subsequent query(...) function will talk to the local metadata database.
Now let’s do it manually without any R function calls.
The default AnnotationHub is at https://annotationhub.bioconductor.org/. By visiting the page we can find several relevant endpoints:
/metadata/annotationhub.sqlite3/fetch/:id # id => rdatapaths.id
So as long as we get the rdatapaths.id of the EnsDb using the metadata, we can download it via the /fetch/:id endpoint.
After downloading the metadata database https://annotationhub.bioconductor.org/metadata/annotationhub.sqlite3, we can inspect it in SQLite3 by connecting it directly:
sqlite3 annotationhub.sqlite3
Some useful commands to inspect a foreign database (or the ultimate help command .help):
sqlite> .header on
sqlite> .mode column
sqlite> .tables
biocversions rdatapaths schema_info test
input_sources recipes statuses timestamp
location_prefixes resources tags
sqlite> .schema rdatapaths
CREATE TABLE `rdatapaths`(`id` integer DEFAULT (NULL) NOT NULL PRIMARY KEY , `rdatapath` varchar(255) DEFAULT (NULL) NULL, `rdataclass` varchar(255) DEFAULT (NULL) NULL, `resource_id` integer DEFAULT (NULL) NULL, `dispatchclass` varchar(255) DEFAULT (NULL) NULL, CONSTRAINT `rdatapaths_ibfk_1` FOREIGN KEY (`resource_id`) REFERENCES `resources`(`id`));
CREATE INDEX `rdatapaths_resource_id` ON `rdatapaths` (`resource_id`);
So let’s make a SQL query to find all Human’s EnsDb:
SELECT r.ah_id, rdp.id AS rdatapaths_id, rdp.rdatapath, r.title
FROM resources AS r
JOIN rdatapaths AS rdp
ON r.id = rdp.resource_id
WHERE r.title LIKE '%EnsDb for Homo Sapiens%';
-- ah_id rdatapaths_id rdatapath title
-- ---------- ------------- -------------------------------------- -- ---------------------------------
-- AH53211 59949 AHEnsDbs/v87/EnsDb.Hsapiens.v87.sqlite Ensembl 87 EnsDb for Homo Sapiens
-- AH53715 60453 AHEnsDbs/v88/EnsDb.Hsapiens.v88.sqlite Ensembl 88 EnsDb for Homo Sapiens
-- AH56681 63419 AHEnsDbs/v89/EnsDb.Hsapiens.v89.sqlite Ensembl 89 EnsDb for Homo Sapiens
-- AH57757 64495 AHEnsDbs/v90/EnsDb.Hsapiens.v90.sqlite Ensembl 90 EnsDb for Homo Sapiens
All the Ensembl releases 87+ are available! I will use the release 90 for example. we can download it by its rdatapaths id:
wget -O EnsDb.Hsapiens.v90.sqlite https://annotationhub.bioconductor.org/fetch/64495
For older Ensembl release, one may need to build the SQLite database based by the instructions from ensembldb. For the last GRCh37 release, Ensembl release 75, one can download the source of the Bioconductor annotation package EnsDb.Hsapiens.v75 and extract it. The database will be under inst/extdata.
Manual query in EnsDB
EnsDb SQLite database are Ensembl annotation databases created by the R package ensembldb.
Here I will show how to find a transcript’s gene name, its genomic location, and all its exon locations given its Ensembl transcript ID.
First connect the database by sqlite3 EnsDb.Hsapiens.v90.sqlite. Its table design is very straightforward:
sqlite> .tables
chromosome exon metadata protein_domain tx2exon
entrezgene gene protein tx uniprot
So it didn’t take me long to figure out how to join the transcript and gene information:
SELECT tx.tx_id, tx.gene_id, gene.gene_name, seq_name, seq_strand
FROM tx JOIN gene ON tx.gene_id = gene.gene_id
WHERE tx_id='ENST00000358731';
-- tx_id gene_id gene_name seq_name seq_strand
-- --------------- --------------- ---------- ---------- ----------
-- ENST00000358731 ENSG00000145734 BDP1 5 1
And for the genomic ranges of its exon:
SELECT tx_id, exon_idx, exon_seq_start, exon_seq_end
FROM tx2exon JOIN exon ON tx2exon.exon_id = exon.exon_id
WHERE tx_id = 'ENST00000380139'
ORDER BY exon_idx;
-- tx_id exon_idx exon_seq_start exon_seq_end
-- --------------- ---------- -------------- ------------
-- ENST00000380139 1 32427904 32428133
-- ENST00000380139 2 32407645 32407772
-- ENST00000380139 3 32407250 32407338
-- ENST00000380139 4 32404203 32404271
-- ENST00000380139 5 32400723 32403200
All the coordinates are 1-based and the ranges are inclusive.
Summary
By downloading the underlying annotation database, one can do the same annotation query out of R language and sometimes it may be helpful. I feel like instead of trying to come up with my own layout of annotation mapping across multiple sources, it is more reliable to use a more official build. On the other hand, it is very hard to get the annotation mapping correct and there are tons of corner cases that require careful and systematic decisions. So I don’t really recommend to build my own mapping at the first place anyway. The method here should help the situation of annotation query out of R a bit.
Potentially one can try copy the full R infrastructure but using the same underlying database and replicate the same experience to other languages, but it might require substantial work to get the infrastructure done and correct.
EDIT 2017-12-13: Add instructions of using older Ensembl release.
EDIT 2019-01-29: Add the web interface of AnnotationHub.
-
Based on my impression, my R expert friends would probably recommend me to write it with R-cpp, which I think would be over-kill for such a small task. But my impression can be wrong. Feel free to share your thoughts! ↩