ID cross reference with exact protein sequence identity using UniParc
TL;DR: Many existing ID mappings between different biological ID systems (RefSeq/Ensembl/UniProt) don’t consider if the IDs have the same exact protein sequence. When the exact sequence is needed, UniParc can be used to cross-reference the IDs. I will demonstrate how to use UniParc to map RefSeq human proteins to UniProt and Ensembl at scale.
You can skip to the solution if you already know what’s the problem I want to tackle.
Camps of biological IDs
There are a few “camps” of biological IDs that are used by many (human) databases and datasets: Ensembl, RefSeq (plus NCBI/Entrez Gene), and UniProt. Each ID camp is comprehensive independently, containing gene-level, transcript-level, and protein-level information using their own systems of IDs. Unfortunately, all three ID systems/camps are useful in their own way, making the choice of the “favorite” ID system really divided for different databases and datasets.
To get a sense of these “ID camps” and how information is connected through them and across them, this great illustration from bioDBnet sums it all (it’s huge):
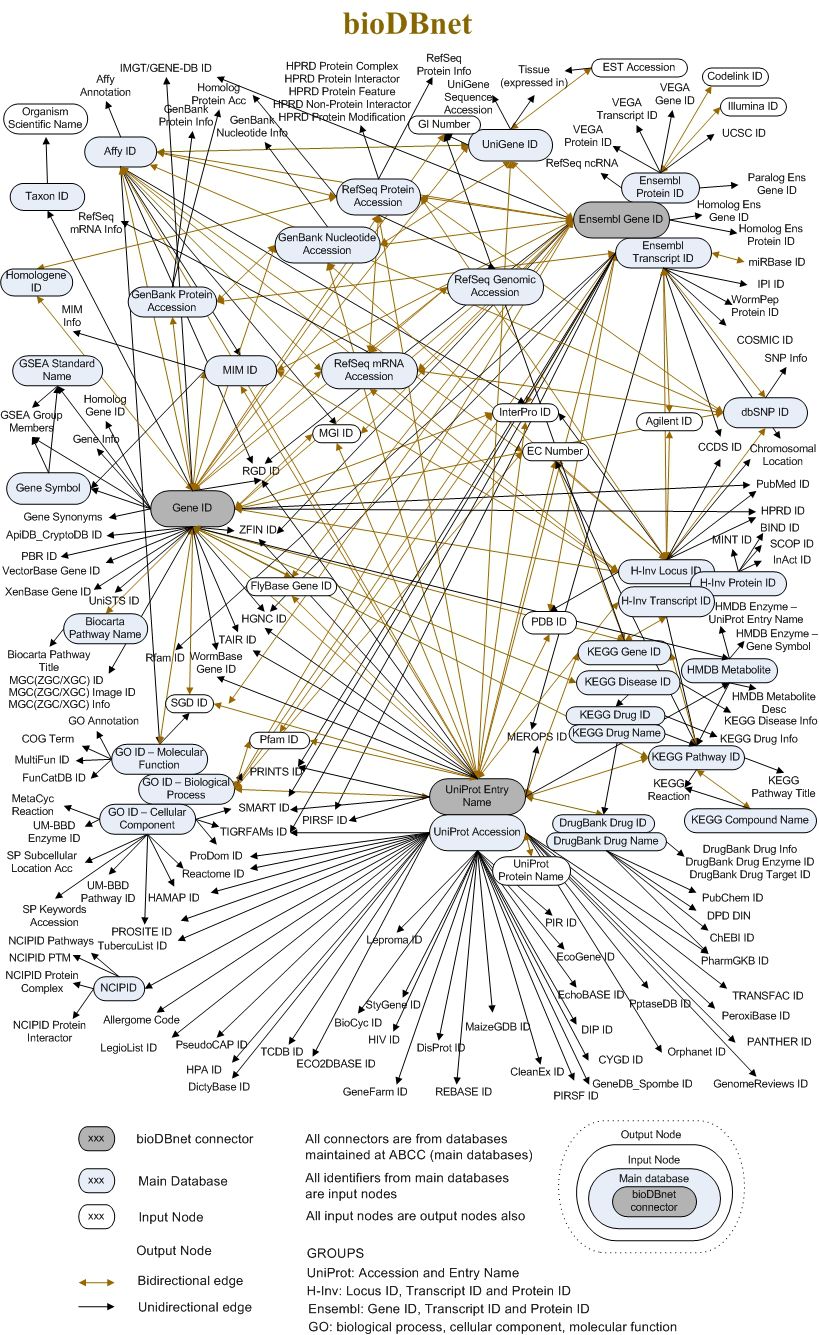
Challenges to map IDs with exact sequence identity
It’s usually straightforward to cross reference within each ID camp, as long as one has the versioned ID and a copy of that camp’s ID system. For example, to know the gene symbol of ENSP00000368632.3, I can easily use ensembldb, a lite copy of Ensembl’s ID system, to find out it is translated from transcript ENST00000379328.9, which is one of the transcripts of gene ENSG00000107485.18, whose gene symbol is GATA3. Easy peasy. The story goes the same for RefSeq and UniProt (albeit this one is more protein centric).
However, things get messy when one wants to cross reference across ID camps. While both official and third-party services (e.g., bioDBnet and DAVID) exist, they don’t guarantee sequence identity match. In this post, I will focus on the protein sequence. For example, bioDBnet says ENSP00000368632 can be mapped to (note the lack of version):
- RefSeq: NP_002042, NP_001002295, …
- UniProt: P23771, …
But when considering their sequence being identical, ENSP00000368632.3 should only match NP_001002295.1 in RefSeq, and the non-canonical form of UniProt P23771-2 (sequence version 1). The other IDs don’t have the exact same protein sequence because of an 1aa deletion. To complicate things more, many mappings don’t handle ID versions, and sequence often changes across ID versions. Without tracking the versioned ID, it’s impossible to say which IDs have the same sequence.
Let me be clear that there is nothing wrong about these services. They’re built so people can map IDs at high level and increase the number of mappable IDs, which is extremely useful in its own way. In fact, a lot of IDs simply cannot be mapped when consider exact sequence identity.
For transcripts the situation isn’t any better because many mapped transcripts of different ID camps have different UTR sequences. I will probably write another post to touch on transcript ID mapping. The short answer is that the MANE project started by RefSeq and Ensembl is working on the problem.
Why mapping with exact sequence identity?
Well, when do I need ID mapping with exact protein sequence identity? My use case is to map post-translation modifications (PTMs) from RefSeq to UniProt. CPTAC detected loads of PTMs (mostly phosphosites) using RefSeq as the peptide spectral library (peptide search database). But a lot of what we know about a protein is from UniProt. So I need a reliable way to map a specific amino acid of one RefSeq protein to its UniProt counterpart. I’ve been using existing services for the job, but they not perfect.
For example, we found a phosphosite NP_001317366.1 (PTPN11) p.Y546. PTPN11 corresponds to Q06124 in UniProt reviewed proteome (the current canonical isoform is Q06124-2 seq. ver3). But you won’t find anything (e.g. antibody) about this site at 546aa because sequences of NP_001317366.1 and Q06124-2 don’t match. This phosphosite actually maps to Q06124-2 p.Y542.
While one can argue that I should re-run the peptide search using UniProt, this solution only works around the problem. The problem comes back when I want to map the Ensembl based mutations to UniProt. I am also aware that two proteins with different sequence can be biologically different, and we shouldn’t just blindly integrate their annotation. I totally agree, so the integration should be further validated. Due to the nature of the shotgun proteomics, as long as the peptide sequence of the PTM site can be found in both proteins, it’s fairly possible that the site can be mapped to both. This topic has been on my mind for a while. I’ll write about it once I figure out the details. Anyway, mapping PTMs between different protein sequences is my next step, and it goes beyond the scope of this post.
UniParc comes into rescue for protein sequence identity
The UniProt Archive (UniParc) is a comprehensive and non-redundant database that contains most of the publicly available protein sequences in the world. Proteins may exist in different source databases and in multiple copies in the same database. UniParc avoided such redundancy by storing each unique sequence only once and giving it a stable and unique identifier (UPI) making it possible to identify the same protein from different source databases. A UPI is never removed, changed or reassigned. UniParc contains only protein sequences.
(source: UniParc help page)
UniParc is a collection of non-redundant protein sequence archive. The UniParc ID and sequence is permanently stable, but the cross-references associated to one UniParc entry may change over time. All of its properties make UniParc perfect to be the identifer to map across ID camps. UniParc IDs can be queried using the CRC64-ISO checksum of the protein sequence.
For example, let’s find the UniPrac ID of NP_001317366.1. First, we obtain its protein sequence in FASTA from NCBI:
$ export refseq_id="NP_001317366.1"
$ curl -Lo "$refseq_id".fasta \
"https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=protein&id=$refseq_id&rettype=fasta&retmode=text"
$ head -n 3 "$refseq_id".fasta
>NP_001317366.1 tyrosine-protein phosphatase non-receptor type 11 isoform 3 [Homo sapiens]
MTSRRWFHPNITGVEAENLLLTRGVDGSFLARPSKSNPGDFTLSVRRNGAVTHIKIQNTGDYYDLYGGEK
FATLAELVQYYMEHHGQLKEKNGDVIELKYPLNCADPTSERWFHGHLSGKEAEKLLTEKGKHGSFLVRES
Then calculate the CRC64 checksum (there are a few packages capable, or use EBI’s checksum calculator):
>>> from pysam import FastaFile
>>> from crc64iso import crc64iso
>>> fa = FastaFile('NP_001317366.1.fasta')
>>> seq = fa.fetch(fa.references[0])
>>> crc64iso.crc64(seq)
'37E8BFC7ECA2D03F'
Search checksum:37E8BFC7ECA2D03F on UniParc gives an unique entry1 UPI000041C017: https://www.uniprot.org/uniparc/?query=checksum%3A37E8BFC7ECA2D03F&sort=score&direct=yes.
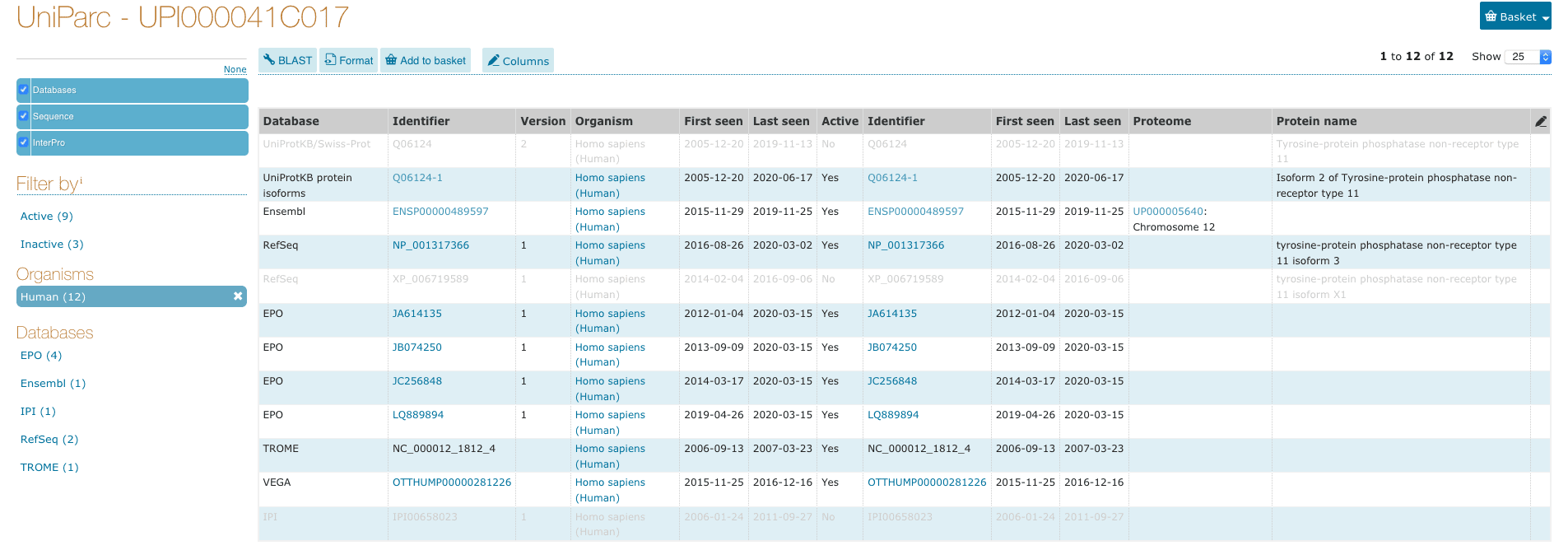
All the external IDs listed here have the identical protein sequence to NP_001317366.1, which of course includes itself. UniParc also marks the IDs inactive if they are superseded by a newer version or become obsolete, which is quite useful for data forensics.
Programatic UniParc access using its XML
To extract UniParc’s cross reference, it’s easiest to parse its XML, which is also easy to download in bulk.
Continue to use UPI000041C017 (NP_001317366.1) as the example,
$ curl -LO https://www.uniprot.org/uniparc/UPI000041C017.xml
$ head -n 10 UPI000041C017.xml
<?xml version='1.0' encoding='UTF-8'?>
<uniparc xmlns="http://uniprot.org/uniparc" xmlns:xsi="http://www.w3.org/2001/XMLSchema-instance" xsi:schemaLocation="http://uniprot.org/uniparc http://www.uniprot.org/docs/uniparc.xsd" version="2020_03">
<entry dataset="uniparc">
<accession>UPI000041C017</accession>
<dbReference type="UniProtKB/Swiss-Prot" id="Q06124" version_i="2" active="N" version="2" created="2005-12-20" last="2019-11-13">
<property type="NCBI_GI" value="84028248"/>
<property type="NCBI_taxonomy_id" value="9606"/>
<property type="protein_name" value="Tyrosine-protein phosphatase non-receptor type 11"/>
<property type="gene_name" value="PTPN11"/>
</dbReference>
While I don’t find XML easy to read, I’ve figured out a way before to parse XMLs as a JSON-like dictionary given its schema. Let’s define a function to flatten the nested structure and only select the information we want:
def parse_dbref(entry):
"""
Parse Ensembl/UniProt/RefSeq IDs of an UniParc entry.
Keep both active and inactive IDs.
"""
ref_ids = {}
for db_type in ["Ensembl", "UniProt", "RefSeq"]:
ids = set()
for d in entry['dbReference']:
if d["@type"].startswith(db_type):
# Skip non-human entries
ncbi_taxid = next(
(p['@value'] for p in d['property'] if p['@type'] == 'NCBI_taxonomy_id'),
None
)
if ncbi_taxid != '9606':
continue
# Make versioned ID
if '@version' not in d:
# Use the UniParc internal version (for UniProt)
id_str = f"{d['@id']}.{d['@version_i']}"
else:
id_str = f"{d['@id']}.{d['@version']}"
ids.add(id_str)
ref_ids[db_type] = sorted(ids)
return ref_ids
xmlschema makes it really easy to parse a XML with schema:
>>> import xmlschema
>>> from pprint import pprint
>>> xs = xmlschema.XMLSchema('https://www.uniprot.org/docs/uniparc.xsd')
>>> data, errors = xs.to_dict('UPI000041C017.xml', validation='lax')
>>> pprint(parse_dbref(data['entry'][0]))
{'Ensembl': ['ENSP00000489597.1'],
'RefSeq': ['NP_001317366.1', 'XP_006719589.1'],
'UniProt': ['Q06124-1.1', 'Q06124.2']}
Voilà, we can now map across ID camps with confidence!
This method can be applied to a large number of queries efficiently. By reading in a FASTA of protein sequences of interest, we can build URLs to UniParc XML per protein entry using its checksum, and pass the URLs as an aria2's input file xml.links:
https://www.uniprot.org/uniparc/?query=checksum%3A{crc64_checksum}&format=xml
out={protein_id}.xml
...
And download all the links in batch:
aria2c -c -j5 --max-overall-download-limit=10M -i xml.links
Summary
We solve the ID mapping with exact protein sequence identity between Ensembl/RefSeq/UniProt camps through UniParc.
Note that the version of UniProt entries is a bit confusing. For example, Q06124.2 means the sequence version 2 of Q06124. But finding UniProt’s sequence version is not that straightforward, and the UniProt isoforms unlike the canonical isoform lack version tracking. As a result, while processing UniProt associated annotation, I will always add UniParc IDs or keep its protein sequence for future reference.
RefSeq and Ensembl protein IDs are always versioned. Thus it’s highly recommended to keep the versioned ID for these two systems.
-
It’s possible that two different protein sequences have the same checksum, though very unlikely. So always double check so do this in batch. ↩